Program Leader: Janet Wiles
Research in the GRN program tackled fundamental questions about growth and form in cellular biology. In this program, computational modelling was used to study how the control of development results from an interaction between each cell's genetic regulatory network and its inputs from neighbouring cells and its environment, and how the process proceeds reliably, while coping with unreliable components, perturbation, injury, and changing environments.
Projects:
- Insights from modelling RNA networks
- Practical software engineering techniques for regulatory models in biology
- Modelling regulatory networks at cell, tissue and organism level
- Modelling GRNs: From complex systems to systems biology
- eScience and complex systems: Developing intuitive Web interfaces to complex simulation algorithms
- Modularity in genetic networks
- An integrated approach to multicellular development
- Case studies in dynamics of complex systems
- A visual framework for online biological sequence analysis
- Radial basis function neural networks with recursive bias field correction
- Virtual drug discovery
- Modelling globular cell colony growth
- Modelling and mapping genes for complex phenotypes in humans
- Patterns in complex systems models
- On the training of mixture of experts with multilevel gating and expert networks
- Kernel methods for biological sequence analysis
- Developmental complexity and bias: modelling and visualisation
- Modelling ontogenetic control points
- Applying complex systems models to muscle-specific gene
- Machine learning architectures for biological sequence analysis
- Modelling gene regulatory networks and plant morphology
- General GRN publications
Insights from modelling RNA networks
Project Leader: Janet Wiles
Researchers: Kevin Burrage, Jennifer Hallinan, John
Mattick
One of the most exciting new ideas for understanding the regulation of gene expression involves the contribution of intronic and other non-protein coding RNAs to regulatory networks within cells. This novel role for intronic RNA is currently making headlines within the molecular biology community but has not yet been modelled computationally. The network of genetic regulatory interactions forms a complex system which is amenable to computational analysis. This project aimed to extend current models to incorporate intronic RNA feedback control, complementing parallel studies in vivo, and computationally testing ideas essential to the theoretical understanding of the basis of life.
In 2004, the research team developed a number of mechanisms for fast decomposition of problems. The outcomes of this research were a paper published in LNCS, AI 2004 conference and two other papers under review.
- Geard, N., Wiles, J., "A gene network model for developing cell lineages", to appear in Alife Journal, 2005.
- Hallinan, J., Wiles, J., "Asynchronous dynamics of an artificial genetic regulatory network", Artificial Life IX: Proceedings of the Ninth International Conference on the Simulation and Synthesis of Living Systems, 2004, 399-403.
- Hallinan, J., Wiles, J., "Evolving genetic regulatory networks using an artificial genome", Proc. Second Asia-Pacific Bioinformatics Conference (APBC2004), 2004; Conferences in Research and Practice in Information Technology, Vol. 29, 291-296.
- Watson, J., Geard, N., Wiles, J., "Toward more biological mutation operators in gene regulation studies", Biosystems, Vol. 76, No. 1-3, 2004, 239-248.
Practical software engineering techniques for regulatory models in biology
Project Leader:
Janet Wiles
Researchers:
Jim Hanan, James Watson
This project investigated software engineering techniques that can be incorporated into the development and use of regulatory models in biology. Through an online survey and interviews with members of the biological modelling community, key characteristics of regulatory model development were identified, and techniques that can address the unique needs of such modellers were recommended.
In 2004, data gathered from the regulatory modelling community through an online survey and focused interviews led to the identification of four key characteristics of the simulations used within this community. These were small team sizes, transient systems, rapidly evolving specifications and non-linear behaviour. Software engineering techniques that cater for these characteristics were identified. These techniques fall into three categories of recommendations: the maintenance of static components, tracking of component interactions, and visualisation techniques that aid understanding of emergent interactions.
- Watson, J., Abbass, H., Lokan, C., Lindsay, P., "Software engineering for artificial life, complex systems, and agent based distillation", Proceedings of the 7th Asia-Pacific Complex Systems Conference, 2004, 649-661.
Modelling regulatory networks at cell, tissue and organism level
Project Leader: Jim Hanan
Researchers: Kevin Burrage, Robert McLeay, Tim Rudge, Janet Wiles
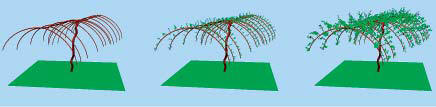
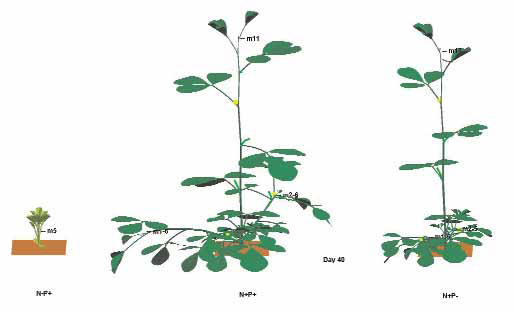
Investigations have been undertaken at the three different scales of biological systems in 2004. Extension of the cell layer modelling system, Cell Modeller, allows improved solution of dynamical systems. A new algorithm for polar cell behaviour has been implemented to further collaborative work with Jim Haselhoff, University of Cambridge, UK. An L-system-based prototyping system capturing intra- and inter-cellular signalling has been developed. This incorporates an interface to the Qu-Prolog logic programming language, which will form the basis for further exploration of Belief-Desire-Intention agent-based systems for modelling genetic regulatory networks. In collaboration with Peter Greshoff of the ARC Centre of Excellence for Integrative Legume Research, a prototype model of auto regulation of nodulation in legumes incorporating root-shoot signalling at whole plant scale has been developed.
In 2005, at the cellular level, in a joint project with the ARC Centre in Bioinformatics, Robert McLeay developed hierarchical Petri Net models of macrophage regulatory and signalling networks. At the intracellular level, Denis Bauer built a Petri Net model of the GRN controlling differentiation in vulval tissues of C. elegans. An invited presentation was given at the International Botanical Congress on the model of genetic control of branching in pea being developed in collaboration with L. Dun and C. Beveridge. During Prof Prusinkiewicz' visit, prototype models were refined for studying genetic control of carbon allocation in plants with Alla Seleznyova, New Zealand HortResearch. (Included 2005/06 summer project)
In 2006, the model of autoregulation of nodulation developed in collaboration with the ARC Centre of Excellence in Integrative Legume Research (CILR) was presented at international conferences in Brisbane, Adelaide and Holland. Carlos Espinosa from the University of Mexico visited, collaborating on genotype phenotype modelling. A prototype model integrating a Petri net simulation system with L-systems visualisations has been completed, in collaborative work with a student intern at the ARC Centre in Bioinformatics. A presentation was made at the ComBio 2006 Conference on modelling of signals involved in regulatory systems of plants.
In 2007, Jim Hanan supervised Frijtof Heyde, a student intern from Germany who implemented a system for modelling spatial and regulatory aspects of intracellular processes-based on an approach integrating Petri Net and L-system models. He also supervised a student, Michael Rivoire, intern from France, who implemented an individual-based model of competition between trees, based on fi eld of neighbourhood concepts. Also in 2007, models of genetic control of pea branching were analysed using Geoff Dromey’s Behavior Tree approach.
- Bian, R., Hanan, J., Chiba, N., "Statistical data directed evolution of L-system models for botanical trees", Proceedings of the 4th International Workshop on Functional-Structural Plant Models, 2004, 253-256.
- Birch, C., Hanan, J., Thornby, D., 'Architectural modeling of maize under water stress', Australian Journal of Experimental Agriculture, Vol. 48, No. 3, 2008, 335-341 .
- Bucciarelli, B., Hanan, J., Palmquist, D, Vance, C.,'A standardized method for analysis of Medicago truncatula phenotypic development', Plant Physiology, .Vol. 142, 2006, 207-219.
- Dun, E.A., Hanan, J., Beveridge, C., “Hypothesisdriven computational modelling of branching control in pea”, Proceedings of the 5th International Workshop on Functional Structural Plant Models , November 2007.
- Godin, C., Hanan, J., Kurth, K., Lacointe, A., Takenaka, A., Prusinkiewicz, P., DeJong, T., Beveridge, C., Andrieu, B., Proceedings of the 4th International Workshop on Functional Structural Plant Models, UMR AMAP, 2004.
- Han, L., Gresshoff, P., Hanan, J., “Virtual soybean—a computational model for studying autoregulation of nodulation”, Proceedings of the 5th International Workshop on Functional-Structural Plant Models , November 2007.
- Hanan, J., Bucciarelli, B., Vance, C., 'Sharing phenotypic data: A coding system and a developmental model', The Medicago Truncatula Handbook, 2006.
- Hanan, J., Thornby, D., Adkins, S., ‘Modelling cotton plant development with L-systems: a template model for incorporating physiology.’, Proceedings of ModSim 05, 2005, 1243–1250.
- Manson, D., Hanan, J., Hunt M., Bristow, M., Erskine,P., Lamb, D., Schmidt, S.'Modelling predicts positive and negative interactions between three Australian tropical tree species in monoculture and binary mixture', Forest Ecology and Management, Vol. 233, No. 2-3,2006, 315-323.
- Renton, M., Hanan, J., "A canonical toolkit for modelling plant function", Proceedings of the 4th International Workshop on Functional-Structural Plant Models, 2004, 226-230.
- Renton, M., Hanan, J., Burrage, K., ‘Using the canonical modelling approach to simplify the simulation of function in functional-structural plant models’, New Phytologist, vol. 166, no. 3, June 2005, 845–857.
- Renton, M., Kaitaniemi, P., Hanan, J., ‘Functional structural plant modelling using a combination of architectural analysis, L-systems, and a canonical model of function.’, Ecological Modelling, vol. 184, no. 2–4, June 2005, 277–298.
- Renton, M., Thornby, D., Hanan, J., 'Canonical modelling: An approach for intermediate level simulation of carbon allocation in functional structural models', Functional-Structural Plant Modelling in Crop Production, Wageningen UR Frontis Series, Springer Verlag, Vol 22, 2007.
- Song, Y., Birch, C., Hanan, J., “Architectural analysis and modeling of maize growth and development under water stress”, Proceedings of the 5th International Workshop on Functional Structural Plant Models, November 2007.
- Stolk, H., Hanan, J., “Discovering genetic regulatory network models in Pisum sativum”, International Congress on Modelling and Simulation (MODSIM 2007), December 2007.
- Thornby, D., Hanan, J., "A modelling exploration of branch extension, defoliation responses, and carbohydrate physiology in cotton", Proceedings of the4th International Workshop on Functional-Structural Plant Models, 2004, 319-322.
- Thornby, D., Spencer, D., Hanan, J., Sher, A., “LDONAX, a growth model of the invasive weed species, Arundo donax L.”, Aquatic Botany , Vol. 87, No. 4, November 2007, 275-284.
- Watanabe, T., Hanan, J., Hasegawa, T., Nakagawa,H., Takahashi, W., Room, P., ‘Rice morphogenesis and plant architecture: Measurement, specification and the reconstruction of structural development by 3D architectural modelling’, Annals of Botany, vol. 95, 2005,
Modelling GRNs: From complex systems to systems biology
Project Leaders: Jennifer Hallinan, Janet Wiles
Researcher: Nic Geard
This project consisted of the production of an extensive literature review on modelling genetic regulatory networks, with an emphasis on methodologies from complex systems science. The project produced a technical report, which is available in both PDF and web format, as well as accompanying seminar materials, all of which are available from: ww.itee.uq.edu.au/~nic/_accs-grn/
The completion of this project in 2004 resulted in: (a) the production of a comprehensive review of the literature on the modelling of gene regulatory networks, which was published as a technical report (b) the production of accompanying seminar / tutorial material; (c) the presentation of a seminar to the ACCS community, and;(d) the presentation of a seminar / tutorial as a guest lecture for the 4th year Cognitive Computing course.
eScience and complex systems: Developing intuitive Web interfaces to complex simulation algorithms
Project Leader: Kevin Burrage
Researchers: Janet Wiles, Roger Sidje, Rachit Srivastava,
Stephen Jeffrey, Tianhai Tian
The project investigated Web technologies that can be used to construct graphical interfaces to complex simulation algorithms. Web standards such as Math ML were used to create user-friendly interfaces to grid computing resources. Simple and intuitive interfaces allow users to run simulations without requiring an expert knowledge of the underlying algorithm. World Wide Web interfaces have been developed for two parallel simulation algorithms: evolutionary rewiring and chemical kinetic simulation in genetic regulation.
In 2004 a prototype interface was constructed and demonstrated using a parallel algorithm for chemical kinetic simulation in genetic regulation. The interface is written in PHP and JavaScript, and is coupled to a MySQL database for storing simulation data, results and parameters. Users access the facility via a login screen and may then proceed to define and execute new simulations or view the results of previous simulations. A suite of administrative tools are provided so that users can actively manage their portfolio of simulations, and users with administrative rights can create, modify and delete user accounts.
Modularity in genetic networks
Project Leader: David Green
Researchers: Tania Bransden, David Cornforth, Alex Heng, David Newth,
Suzanne Sadedin
Biomolecular studies point increasingly to the importance of modularity in the organisation of the genome. Processes such as the maintenance of metabolism are controlled by suites of genes that act as distinct, self contained units or modules. This project used simulation models to investigate the role of processes such as feedback and natural selection that are involved in the emergence of genetic modularity. In his 1986 book, The Blind Watchmaker”, Richard Dawkins used the example of a child trying to type a line from Shakespeare to illustrate evolution. In "Weasel World…", Cornforth and Green extended this idea to open-ended evolution within an artificial universe.
The results achieved in 2004 highlight the role of genetic modularity in allowing the emergence of ever more complex forms. In other work this year, David Green's book The Serendipity Machine included an overview of recent genomic research and a simulation study by Whigham and Green investigated the role of genetic tradeoffs in the differentiation of genotypes.
In 2005 we implemented Weasel World, a model that allows experimentation with the factors that lead to the combination of modules, or building blocks, to form hierarchical structures. With this model, we quantified the effects on genetic module formation of variable length genotype, variable genotype to phenotype mapping, pseudo-spatial environment, and memetic evolution. We showed that information shared between organisms allows them to combine and build larger modules. These results have implications for the understanding of the formation of hierarchical structures in evolution.
Another 2005 achievement is that we used a spatially explicit model of population genetics in a landscape to investigate the effects of genetic trade-offs (where organisms must split resources between different features) on the response of monoecious diploid populations to environmental gradients. One trade-off related the plasticity of an individual to its overall fitness, where individuals that can tolerate a wide range of environmental values are less fit than individuals with a narrow tolerance range. The second trade-off assumed that individuals that can live at in extreme environments are placed under stress and therefore reach maturity later in their life cycle. In both cases the resulting population distributions produced clear genetic differentiation, showing that simple trade-off mechanisms are one route
- Cornforth, D., Green, D., Awburn, J., ‘The formation of hierarchical structures in a pseudo-spatial co-evolutionary artificial life environment’, in Recent Advances in Artificial Life, Advances in Natural Computation-Vol. 3, World Scientific Publishers, 2005, 55–68.
- Cornforth, D., Green, D., Awburn, J., "Weasel world: A simple artificial environment for investigating open-ended evolution", Proceedings of the 8th Asia Pacific Symposium on Intelligent and Evolutionary Systems,2004, 40-49.
- Green, D., Sadedin, S., ‘Interactions matter - Complexity in landscapes and ecosystems’, Ecological Complexity, vol. 2, no. 2, 2005, 117–130.
- Whigham, P., Green, D., "A spatially-explicit model of genetic trade off", Proceedings of the 7th Asia-Pacific Complex Systems Conference, 2004, 91-100.
An integrated approach to multicellular development
Project Leader:
Janet Wiles
Researchers:
Nic Geard, Jim Hanan, Jim
Haseloff, Peter Lindsay, Tim Rudge
In this project, we were concerned with the processes in plants and animals that coordinate the formation of shape. How does a single egg or seed cell develop into the shapes that make up an embryo or adult organism? How does it do this so reliably, even in a noisy environment? The key is the interaction of the signals that are passed between cells and the DNA processing (or genetic regulation) that occurs within each cell. We used complex systems models to study the nature of these interactions and how they control shape formation in a robust way. This analysis allowed us to generalise principles from biological shape formation to apply to the understanding and design of systems in the engineering domain.
- Rudge, T., Geard, N., ‘Evolving gene regulatory networks for cellular morphogenesis’, Advances in Natural Computation - Vol. 3 Recent Advances in Artificial Life, 2005, 231–252.
- Rudge, T., Geard, N., 'Control and constraint: Cross centre insights from modelling cellular morphogenesis', ACCS Technical Report, No. ACCS-TR-06-01, ARC Centre for Complex Systems, February 2006.
- Rudge, T., Haseloff, J., ‘A computational model of cellular morphogenesis in plants’, Advances in Natural Computation - Vol. 3 Recent Advances in Artificial Life, 2005, 78–87.
Case studies in dynamics of complex systems
Project Leader:
Janet Wiles
Researcher:
Shev Macnamara
This project consisted of a tutorial review of the common mathematical techniques underlying the complex systems analyses of current network-based models of RNA. (Summer Project 2003/04)
A visual framework for online biological sequence analysis
Project Leader:
Mikael Boden
Researcher:
Mark Wakabayashi
The currently most successful algorithms for biological sequence analysis employ intricate dynamics and offer powerful prediction tools. In this project we aimed to tap on this power by making our own algorithms available over the internet where their full benefit can be evaluated. Outcomes - consisting of a visual interface and tools - were realised by using our protein subcellular localisation predictor, the Protein Prowler http://pprowler.imb.uq.edu.au.
In 2004, the Java servlet/JSP framework was investigated and used to build visualisation tools and sequence data support tools. These tools are currently being integrated into the Protein Prowler. The project has, besides providing the research assistant and the group with experience in utilising JSP/Servlet technology for sequence analysis also allowed the research assistant to generate new data sets to be used in further development of new prediction modules. A research paper has been submitted for publication. (Summer Project 2004/05)
- Boden, M., Hawkins, J.,
"Prediction of subcellular localisation using sequence-biased recurrent
networks", to appear in Bioinformatics, 2005.
Radial basis function neural networks with recursive bias field correction
Project Leader:
Geoff McLachlan
Researcher:
Jianxiong Wang
In many important application areas such as control, pattern recognition, and signal processing, nonlinear adaptive systems such as radial basis function (RBF) networks are needed to approximate underlying nonlinear mappings through learning from examples. In this project, we consider the extension of RBF networks to model signal in homogeneities due to acquisition equipments. Such low frequency artifacts (bias field) could be spatial or temporal correlated. By adopting a Markov random field (MRF) model and a recursive bias field correction scheme, the bias field and the true signal intensities can be estimated simultaneously. (Summer Project 2004/05)
Virtual drug discovery
Project Leader:
Peter Adams
Researcher:
Stephen Long
Modern methods in drug discovery are characterised by a large number of candidate drug molecules, often stored in silico. Traditional methods of screening such molecules are inefficient, so computational approaches need to be developed, to enable high-throughput screening. In this project we continued investigation into a method for rapidly identifying structures on the surface of candidate molecules which may indicate a higher probability of identifying lead molecules. (Summer Project 2003/04)
Modelling globular cell colony growth
Project Leader:
Kevin Burrage
Researcher:
David Woolford
This work was motivated by the desire to model the growth pattern of stem cell neural spheres, which start as a single cell and grow to a sphere containing around 16000 cells. The proposed system models globular cell colony growth in a 3D environment using OpenGL. The end result of the project was a program, executable on Linux based systems, that facilitates user interaction and animates cell colony growth starting with a solitary cell.(Summer Project 2003/04)
Modelling and mapping genes for complex phenotypes in humans
Project Leader: Jacqueline Wicks
Researchers: Ananthila Anandacoomarasamy, David Duffy, Katrina
Scurrah, Susan Wilson
The genotype-phenotype correspondence in humans is difficult to uncover for a number of reasons. For complex phenotypes, there may be hundreds of genes that influence the phenotype, as well as environmental, and the more recently discovered epigenetic influences on phenotypes. This project aimed to develop mathematical, statistical and computational approaches to mapping genes that influence phenotypes of interest in humans, and to apply the methods in gene mapping projects currently underway in Australia. The emphasis was on flexible modelling that captured the important influences of complex disease genetics, and allowed for multi-gene, environmental, and epigenetic interactions.
In 2005, several significant papers were finalised and published in high quality journals during the year. Two papers presented results from gene mapping studies, for the complex disease endometriosis, that Dr Wicks was involved in. These were conducted at the Queensland Institute of Medical Research and headed by Dr Susan Treloar. Another paper was published which described new conceptual approaches to gene mapping. In April Dr Wicks was an Invited Discussant at the prestigious International Statistical Institute, and presented a paper on some theoretical results in gene mapping. Several collaborations on future gene mapping projects in Australia were also initiated.
In 2006, a paper was completed on the influence of a particular gene, called the P2X7 polymorphism, on the autoimmune disease systemic lupus erythematosus. A paper on mathematical aspects of disease gene mapping was published. Finally, research was undertaken in the highly topical area of epigenetics in human disease. A method for detecting genes under the influence of imprinting is being developed, and the first stages of the design of software to implement the method are underway.
- Treloar, S., Wicks, J., Nyholt, D., Montgomery, G.,Bahlo, M., Smith, V., Dawson, G., Mackay, I., Weeks,D., Bennett, S., Carey, A., Ewen-White, K., Duffy, D.,O'Connor, D., Barlow, D., Martin, N., Kennedy, S.,‘ Genome wide linkage study in 1 176 affected sister pair families identifies a significant susceptibility locus for endometriosis on chromosome 10q26’, American Journal of Human Genetics, vol. 77, no. 3, 2005, 365–376.
- Treloar, S., Zhao, Z., Armitage, T., Duffy, D., Wicks,J., Martin, N., ‘Association between polymorphisms in the progesterone receptor gene and endometriosis’, Molecular Human Reproduction, vol. 11, no. 9, September 2005, 641–647.
- Wicks, J., ‘Genetic modelling assumptions for gene mapping and the triangle constraints’, Bulletin of the International Statistical Institute, 2005.
- Wicks, J., Treloar, S., Martin, N., Duffy, D.,
‘New concepts for distinguishing the hidden
patterns of linkage disequilibrium which underlie association
between genotypes and complex phenotypes’,
Twin Research
and
Human Genetics, vol. 8, no. 2, April 2005, 95–100.
Patterns in complex systems models
Project Leader: Janet Wiles
Researchers: David Carrington, Jim Hanan, James
Watson, Mark Wakabayashi
The aim of this project was to find and document experience in complex systems modelling using a patterns methodology taken from software engineering. The project had three main goals: identify an initial collection of inter-related GRN patterns with potential for application in all of the Centre's domains, documented in a form comprehensible to all complex systems researchers; establish a framework for a patterns library which allows complex systems researchers to contribute and ratify solutions; and raise awareness in the complex system's community of the value of patterns in advancing the field.
In 2005, an online presence for the project was developed (http://www.itee.uq.edu.au/~patterns/), which includes a submission mechanism for new pattern proposals. A patterns template and classification scheme suitable for complex systems modelling was adapted from software engineering. Software demonstrating the patterns approach was implemented and made available online. Two workshops were held to foster interest in the complex systems community and to develop new patterns through cross-disciplinary collaboration. The first was held on 6–7 June and resulted in successful collaborations between ACCS, UQ’s School of Information Technology & Electrical Engineering, the ARC Centre in Bioinformatics and CSIRO researchers. The second was held 5 December as a special session of the Second Australian Conference on Artificial Life (ACAL'05).Six complex systems modelling patterns were developed, four in collaboration with attendees of the first workshop. A paper describing patterns in complex systems modelling was presented at the IDEAL conference in Brisbane, and published in the Lecture Notes in Computer Science series. A paper discussing the computational modelling process, and developed in collaboration with ITEE and Institute of Molecular Bioscience researchers, was presented in June at GECCO in Washington. Two further papers resulting from discussions at the first workshop were presented at The Second Australian Conference on Artificial Life in Sydney, and published in the Advances in Natural Computation series.
This project led to a summer project in collaboration with the Queensland Brain Institute. The ACCS patterns project provided the infrastructure for collating patterns in complex systems modelling. The summer project involved the development of patterns used within ACCS research, in particular for representing information using tree diagrams. The software patterns developed in the project were used in a software tool to model neurosphere growth. The neurosphere modelling tool enables researchers to specify rules for how each cell divides, and then view how it grows from a single cell to several thousand cells. (Included 2005/06 summer project).
- Geard, N., Willadsen, K., Wiles, J., ‘Perturbation analysis: A complex systems pattern’, Recent Advances in Artificial Life, 2005; Advances in Natural Computation- Vol. 3, 69–83.
- Watson, J., Hawkins, J., Bradley, D., Dassanayake, D., Wiles, J., Hanan, J., ‘Towards a network pattern language for complex systems’, Recent Advances in Artificial Life, 2005; Advances in Natural Computation-Vol. 3, 309–317.
- Wiles, J., Watson, J., ‘Patterns in complex systems modelling’, Proceedings of Sixth International Conference on Intelligent Data Engineering and Automated Learning (IDEAL'05); Lecture Notes in Computer Science, vol. 3578, 532–539.
On the training of mixture of experts with multilevel gating and expert networks
Project Leader:
Geoff McLachlan
Researcher:
Kui Wang
This project studied the methodology in incorporating multilevel data hierarchies within networks modelling. The proposed research aimed at generalizing Mixtures of Expert (ME) networks by allowing both the gating and expert networks to incorporate the multilevel data hierarchy via the generalised linear mixed-effects model. That is, we assumed that each unit contributes (scalar) random effects within the gating and expert networks through the corresponding linear predictors. We also aimed to develop fast learning of the proposed generalised ME network via the EM algorithm. The extension of the methodology to Gaussian mixtures and multi-layered perceptron neural networks for tackling problems with hierarchically structured data was also be studied. Although neural networks and mixtures of experts can be applied to problems in network-based systems in general, the aim here was to apply them to problems in genetic regulatory networks, such as the analysis of time-course gene expression data.
The project started in mid April 2005. We have extended mixture-of-experts networks to incorporate multilevel data hierarchies via the generalised linear mixed effects model. Research papers have been produced to disseminate the research findings to a wide audience in highly-regarded international journals and conferences in relevant scientific fields.
- Ng, S., McLachlan, G., ‘Mixture model-based statistical pattern recognition of clustered or longitudinal data’, Proceedings of WDIC 2005, APRS Workshop on Digital Image Computing, 2005, 139–144.
- Ng, S., McLachlan, G., ‘Normalized Gaussian networks with mixed feature data’, Lecture Notes in Artificial Intelligence, vol. 3809, 2005, 879–882.
Kernel methods for biological sequence analysis
Project Leader: Mikael Boden
Researchers:
Lynne Davis, Johnson Shih
This project developed a flexible software library for kernel methods as applied to data-driven biological sequence analysis. Several example kernels selected from the literature were tested. Benchmarking targeted bioinformatics modelling problems such as organellar protein import and protein subnuclear localisation.(2005/06 summer project)
Developmental complexity and bias: modelling and visualisation
Project Leader:
Janet Wiles
Researcher:
Nic Geard
The development and evolution of biological organisms are highly complex processes. A single egg cell contains in its genome the information to produce an adult form. Evolution, of millions of years, has produced a diverse range of different forms. Biology currently lacks a comprehensive theoretical framework for understanding how development interacts with evolution. Computational modelling is a methodology that, by focusing attention on the core aspects of a complex system, can enable novel insights into its behaviour. The aim of this project was to produce two journal submissions based on Nic Geard's PhD thesis. The first described the interactive visualisation software tools developed to visualise the complexity of model developmental systems. The second reported the insights obtained into the way in which the intrinsic dynamics of a model developmental system can bias its functional behaviour, and the implications this has for the direction of evolution.
In 2006, a modelling and visualisation framework was developed for exploring the interactions that occur between development and evolution. This framework allowed us to visualise both individual cell lineages and how these lineages vary over evolutionary space. The most important theoretical finding that emerged from this research was the manner in which the dynamics of a developmental system can influence the direction of evolution. Furthermore, the use of computational modelling as a methodology enables a rigorous and quantitative approach to investigating and evaluating hypotheses that are otherwise often framed in a general fashion.
Modelling ontogenetic control points
Project Leader: Janet
Wiles
Researcher: James Watson
Biology rarely reinvents the wheel. Evolution has produced points of control, such as hox genes, that enable the effective reuse and adaption of existing biological components, and thus the development and maintenance of increasingly complex organisms. Components from nucleotides through to cells and tissues similarly offer forms of ‘biological abstraction’ that allow the genetic system to control complex biological systems operating in noisy environments. Being abstractions, computational models of GRNs are most effective when they target these points of control. This project had two goals. The first was to identify and model these control points. A software pipeline connecting ontogenetic levels (nucleotide to regulation to morphology) was used as the basis for modelling these mechanisms. The second goal was to investigate the application of these mechanisms, which have been honed by millions of years of evolutionary constraints, to the control of other complex systems across the ACCS's programs.
In 2006, to make existing ACCS GRN software available to a wider computational modelling community, a reusable library of in-house modelling software(named CoolKit) has been initiated. This library is being developed in collaboration with Jared Moore, an ACCS summer scholar. A revision of an existing software suite that ties together computational models of a genetic sequence, regulatory network, plant development, and evolution, has been undertaken to capitalise on existing distributed computing facilities. A 3D model of early sponge development, based on simple interactions between cells and simulated annealing, has been implemented.
Applying complex systems models to muscle-specific gene
Project Leader: Janet Wiles
Researcher: Kai Willadsen
This project aimed to apply complex systems modelling techniques to existing biological problems of interest to industry through collaboration with researchers within CSIRO Livestock Industries. This research focuses on a gene network model for muscle formation in cattle recently developed by CSIRO researchers. Previous research has explored the structure of this network using an evolutionary technique to determine possible extreme phenotypes when Myogenin is inactivated; Myogenin is a muscle-specific transcription factor whose inactivation is known to cause a severe reduction in muscle development. In this project, complex systems-based approaches, including Boolean networks and neural networks, are being used to explore the dynamics and the robustness of this gene coexpression network.
In 2006, the muscle-specific myogenin gene network provided by CSIRO was adapted for simulation using complex-systems modelling techniques. A modelling framework was developed and used to simulate and analyse this network. Methods for dealing with uncertainty and different interpretations of network behaviour were introduced and software support tools for these methods were written. Preliminary results based on the analyses of the framework were presented to CSIRO researchers. Findings on the methodological challenges involved in this project have been recorded in a technical report, which is in the process of being finalised.
Machine learning architectures for biological sequence analysis
Project Leader: Mikael Boden
Researchers: Daniel Bradley, John Hawkins, Janet
Wiles
This project contributed a software technique, and concrete implementation that will allow the exploration of the nuclear portion of the proteome, and hence another avenue for exploring gene regulation. The project aimed to develop a nascent piece of IP developed within the Centre and see it through to commercial reality.
In this project in 2007, we have developed working relationships with a number of experimental biologists and have published the results of joint projects using computational biology to generate hypotheses testable in the laboratory. We have recently developed a method for extracting sequence features from Kernel-based machine learning algorithms which will allow us to produce novel predictions regarding the location of functional regions in biological sequences. This technique will form the hub for a series of further joint investigations into the sequence features responsible for the dynamics of assorted nuclear proteins.
- Hawkins, J., Davis, L., Boden, M., “Predicting nuclear localisation”, Journal of Proteome Research, Vol. 6, No. 4, 2007, 1402-1409.
- Bodén, M., Yuan, Z., Bailey, T.L., 'Prediction of protein continuum secondary structure with probabilistic models based on NMR solved structures', BMC Bioinformatics, Vol. 7, No. 68, 2006.
- Hawkins, J., Mahony, D., Maetschke, S., Wakabayashi, M., Teasdale, R.D., Boden, M., “Identifying novel peroxisomal proteins”, Proteins: Structure, Function, and Bioinformatics, Vol. 69, No. 3, 2007, 606-616.
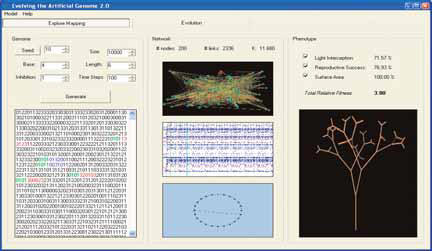
Modelling gene regulatory networks and plant morphology
Project
Leader:
Janet Wiles
Researcher: James Watson
This project had two primary goals. The first was to analyse a computational model of gene regulation and plant morphology. The second was to develop the modelling techniques that allow the efficient simulation and analysis of such a model. In the model of gene regulation and plant morphology, there were three main components - a genetic sequence, a regulatory network, and plant development. This model was analysed with respect to the constraints these components placed on each other. A software toolkit of common modelling functionality was designed, and a prototype regulatory network model that utilised idle, distributed computing resources to reduce simulation time was developed. Other methods of improving simulation time, such as the use of graphics processing units for simulation calculations, were investigated.
In 2007, the analysis of the GRN and plant morphology software model was extended to investigate the constraints different components placed upon each other. An analysis of this model was presented at IPCAT’07in Oxford. A number of research centres, including the San Diego Supercomputer Centre, Calit2, the Centre for Research in Language and the Salk Institute were visited to see first-hand how leading research centres implement and use computational facilities. The software model that utilises idle computing facilities in the student laboratories was outlined in a report to the Queensland Cyber Infrastructure Foundation, and was extended to form a practical parameter sweep tool. This tool was used by researchers from the Institute for Molecular Bioscience, and the results from some of this work were presented at Complex’07 and IPCAT’07. A computational steering workshop (CompuSteer) was attended in Hull, UK, and an overview of this project was presented at the School of Computing, Leeds University.
- Watson, J., Wiles, J., “Modeling the fitness of plant morphologies across three levels of complexity”, Proceedings of the Seventh International Workshop on Information Processing in Cells and Tissues, August2007.
General GRN publications
- Bauer, D.C., Boden, M., Their, R., Yuan, Z., ‘Predicting structural disruption of proteins caused by crossover’, Proceedings of the IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology, November 2005, 514–520.
- Bauer, D., Bodén, M., Thier, R., Gillam, E.M., 'STAR: Predicting recombination sites from amino acid sequence', BMC Bioinformatics, Vol. 7, No. 437, 2006.
- Ben-Tovim J.L., Ng, S., Monico, K., McLachlan, G., "Linking gene-expression experiments with survival-time data.", Statistical Modelling. Proceedings of the 19th International Workshop on Statistical Modelling, 2004, 71-75.
- Bodén, M., Bailey, T.L., 2006 Workshop on Intelligent Systems for Bioinformatics (WISB 2006), Conferences in Research and Practice in Information Technology, Vol. 73, Australian Computer Society, 2006.
- Bodén, M., Bailey, T.L., 'Identifying sequence regions undergoing conformational change via predicted continuum secondary structure', Bioinformatics, Vol. 22, No. 15, 2006, 1809-1814.
- Bodén, M., Hawkins, J., 'Evolving discriminative motifs for recognizing proteins imported to the peroxisome via the PTS2 pathway', 2006 IEEE Congress on Evolutionary Computation, July 2006.
- Boden, M., Hawkins, J., ‘Improved access to sequential motifs: A note on the architectural bias of recurrent networks’, IEEE Transactions on Neural Networks, vol.16, no. 2, 2005, 491–494.
- Coppel, R., Green, D., ‘Database integration and querying in the bioinformatics domain’, Proceedings of the 7th Asia-Pacific Complex Systems Conference, 2005, 14–20.
- Dorr, G., Hanan, J., Adkins, S., Hewitt, A., Noller, B., “Spray deposition on plant surfaces: a modeling approach”, Proceedings of the 5th International Workshop on Functional Structural Plant Models, November 2007.
- Dorr, G., Hanan, J., Woods, N., Kleinmeulmann, P., Adkins, S., Noller, B., 'Simulating spray deposition on plant canopies within a wind tunnel', International Advances in Pesticide Application Conference 2006, January 2006; Aspects of Applied Biology, Vol. 77, 395-403.
- Dorr, G., Noller, B., Hewitt, A., Hanan, J., Adkins, S., 'A decision-making tool to minimise environmental and public health risk of pesticide application', Proceedings 6th Annual Health and Medical Research Conference of Queensland, 2006.
- Dorr, G., Noller, B., Woods, N., Hewitt, A., Hanan, J., Adkins, S., Ricci, P., “Development of a decision-making tool to minimise environmental and public health risk of pesticide application”, Rational Environment Management of Agrochemicals: Risk Assessment, Monitoring, and Remedial Action , American Chemical Society, 2007, 53-65.
- Geard, N., Wiles, J., ‘A gene network model for developing cell lineages’, Artificial Life, vol. 11, no. 1–2,2005, 249–68.
- Geard, N., Wiles, J., “Directed evolution of artificial cell lineages”, Progress in Artifi cial Life , Edited by Randall, M., Abbass, H., Wiles, J., December 2007; Lecture Notes in Computer Science, Vol. 4828, 144-155.
- Geard, N., Wiles, J., “LinMap: Visualising complexity gradients in evolutionary landscapes”, Artificial Life, 2008.
- Hallinan, J., Jackway, P., ‘Network motifs, feedback loops and the dynamics of genetic regulatory networks’, Proceedings of the IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology, 2005.
- Hanan, J., Loch, B., McAleer, T., "Processing laser scanner data to extract structural information", Proceedings of the 4th International Workshop on Functional-Structural Plant Models, 2004, 6-8.
- Hanan, J., Wang, Y., "Floradig: A configurable program for capturing plant architecture", Proceedings of the 4thInternational Workshop on Functional-Structural Plant Models, 2004, 407-411.
- Hawkins, J., Boden, M., ‘The applicability of recurrent neural networks for biological sequence analysis’, IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 2, no. 3, 2005, 243–253.
- Hawkins, J., Boden, M., ‘Predicting peroxisomal proteins’, Proceedings of the IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology, November 2005, 469–474.
- Lohaus, R., Geard, N., Wiles, J., Azevedo, R., “A generative bias towards average complexity in artificial cell lineages.”, Proceedings of the Royal Society of London , Series B, Vol. 274, No. 1619, 2007, 1741-1750.
- Maetschke, S., Towsey, M., Boden, M., ‘BLOMAP: An encoding of amino acids which improves signal peptide cleavage prediction’, Proceedings of the 3rd Asia-Pacific Bioinformatics Conference, 2005, 141-150.
- Myers, B., Dix, McLachlan, G., Chang, S., Mar, J., Ambroise, C., "On the simultaneous use of clinical and microarray expression data in the cluster analysis of tissue samples", Proceedings of the second conference on Asia-Pacific bioinformatics, 2004; ACM International Conference Proceeding Series, Vol. 29, 167-171.T.,
- Stolk, H., Zaluki, M., Hanan, J., “Subpopulation agents emerge from individual agents in simulations of monarch butterflies”, International Congress on Modelling and Simulation (MODSIM 2007), December 2007.
- Suksawatchon, J., Lursinsap, C., Boden, M., ‘Heuristic algorithm for computing reversal distance with multigene families via binary integer programming’, Proceedings of the IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology, November 2005, 187–193.
- Wiles, J., Tonkes, B., ‘Hyperspace geography: Visualising fitness landscapes beyond 4D’, Artificial Life, Special Issue on Visualisation, vol. 12, no. 2, 2006.
- Wiles, J., Watson, J., Tonkes, B., Deacon, T., ‘Transient phenomena in learning and evolution: genetic assimilation and genetic redistribution’, Artificial Life , vol. 11, no. 1–2, 2005, 177–188
- Willadsen, K., Wiles, J., “Robustness and state-space structure of Boolean gene regulatory models”, Journal of Theoretical Biology , Vol. 249, No. 4, 2007, 749-765.